Results
- Showing results for:
- Reset all filters
Search results
-
Journal articleMcCarthy RR, Mazon-Moya MJ, Moscoso JA, et al., 2017,
Cyclic-di-GMP regulates lipopolysaccharide modification and contributes to Pseudomonas aeruginosa immune evasion
, Nature Microbiology, Vol: 2, Pages: 1-10, ISSN: 2058-5276Pseudomonas aeruginosa is a Gram-negative bacterial pathogen associated with acute and chronic infections. The universal cyclic-di-GMP second messenger is instrumental in the switch from a motile lifestyle to resilient biofilm as in the cystic fibrosis lung. The SadC diguanylate cyclase is associated with this patho-adaptive transition. Here, we identify an unrecognized SadC partner, WarA, which we show is a methyltransferase in complex with a putative kinase, WarB. We established that WarA binds to cyclic-di-GMP, which potentiates its methyltransferase activity. Together, WarA and WarB have structural similarities with the bifunctional Escherichia coli lipopolysaccharide (LPS) O antigen regulator WbdD. Strikingly, WarA influences P. aeruginosa O antigen modal distribution and interacts with the LPS biogenesis machinery. LPS is known to modulate the immune response in the host, and by using a zebrafish infection model, we implicate WarA in the ability of P. aeruginosa to evade detection by the host.
-
Journal articleYuan Z, Riera A, Bai L, et al., 2017,
Structural basis of MCM2-7 replicative helicase loading by ORC-Cdc6 and Cdt1
, Nature Structural & Molecular Biology, Vol: 24, Pages: 316-324, ISSN: 1545-9993To start DNA replication, the Origin Recognition Complex (ORC) and Cdc6 load a Mcm2-7 double hexamer onto DNA. Without ATP hydrolysis, ORC-Cdc6 recruits one Cdt1-bound Mcm2-7 hexamer, forming an ORC-Cdc6-Cdt1-Mcm2-7 (OCCM) helicase loading intermediate. Here we report a 3.9Å structure of the OCCM on DNA. Flexible Mcm2-7 winged-helix domains (WHD) engage ORC-Cdc6. A three-domain Cdt1 configuration embraces Mcm2, Mcm4, and Mcm6, nearly half of the hexamer. The Cdt1 C-terminal domain extends to the Mcm6 WHD, which binds Orc4 WHD. DNA passes through the ORC-Cdc6 and Mcm2-7 rings. Origin DNA interaction is mediated by an a-helix in Orc4 and positively charged loops in Orc2 and Cdc6. The Mcm2-7 C-tier AAA+ ring is topologically closed by a Mcm5 loop that embraces Mcm2, but the N-tier ring Mcm2-Mcm5 interface remains open. This structure suggests loading mechanics of the first Cdt1-bound Mcm2-7 hexamer by ORC-Cdc6.
-
Journal articleBosi E, Fondi M, Orlandini V, et al., 2017,
The pangenome of (Antarctic) Pseudoalteromonas bacteria: evolutionary and functional insights
, BMC Genomics, Vol: 18, ISSN: 1471-2164Background:Pseudoalteromonas is a genus of ubiquitous marine bacteria used as model organisms to study the biological mechanisms involved in the adaptation to cold conditions. A remarkable feature shared by these bacteria is their ability to produce secondary metabolites with a strong antimicrobial and antitumor activity. Despite their biotechnological relevance, representatives of this genus are still lacking (with few exceptions) an extensive genomic characterization, including features involved in the evolution of secondary metabolites production. Indeed, biotechnological applications would greatly benefit from such analysis.Results:Here, we analyzed the genomes of 38 strains belonging to different Pseudoalteromonas species and isolated from diverse ecological niches, including extreme ones (i.e. Antarctica). These sequences were used to reconstruct the largest Pseudoalteromonas pangenome computed so far, including also the two main groups of Pseudoalteromonas strains (pigmented and not pigmented strains). The downstream analyses were conducted to describe the genomic diversity, both at genus and group levels. This allowed highlighting a remarkable genomic heterogeneity, even for closely related strains. We drafted all the main evolutionary steps that led to the current structure and gene content of Pseudoalteromonas representatives. These, most likely, included an extensive genome reduction and a strong contribution of Horizontal Gene Transfer (HGT), which affected biotechnologically relevant gene sets and occurred in a strain-specific fashion. Furthermore, this study also identified the genomic determinants related to some of the most interesting features of the Pseudoalteromonas representatives, such as the production of secondary metabolites, the adaptation to cold temperatures and the resistance to abiotic compounds.Conclusions:This study poses the bases for a comprehensive understanding of the evolutionary trajectories followed in time by this peculiar bact
-
Journal articleNanev CN, Saridakis E, Chayen N, 2017,
Protein crystal nucleation in pores
, Scientific Reports, Vol: 7, ISSN: 2045-2322The most powerful method for protein structure determination is X-ray crystallography which relies on the availability of high quality crystals. Obtaining protein crystals is a major bottleneck, and inducing their nucleation is of crucial importance in this field. An effective method to form crystals is to introduce nucleation-inducing heterologous materials into the crystallization solution. Porous materials are exceptionally effective at inducing nucleation. It is shown here that a combined diffusion-adsorption effect can increase protein concentration inside pores, which enables crystal nucleation even under conditions where heterogeneous nucleation on flat surfaces is absent. Provided the pore is sufficiently narrow, protein molecules approach its walls and adsorb more frequently than they can escape. The decrease in the nucleation energy barrier is calculated, exhibiting its quantitative dependence on the confinement space and the energy of interaction with the pore walls. These results provide a detailed explanation of the effectiveness of porous materials for nucleation of protein crystals, and will be useful for optimal design of such materials.
-
Journal articlePearson JS, Giogha C, Muhlen S, et al., 2017,
EspL is a bacterial cysteine protease effector that cleaves RHIM proteins to block necroptosis and inflammation
, NATURE MICROBIOLOGY, Vol: 2, ISSN: 2058-5276 -
Journal articleBernal P, Allsopp LP, Filloux AAM, et al., 2017,
The Pseudomonas putida T6SS is a plant warden against phytopathogens
, The ISME Journal, Vol: 11, Pages: 972-987, ISSN: 1751-7362Bacterial type VI secretion systems (T6SSs) are molecular weapons designed to deliver toxic effectors into prey cells. These nanomachines play an important role in inter-bacterial competition and provide advantages to T6SS active strains in polymicrobial environments. Here we analyse the genome of the biocontrol agent Pseudomonas putida KT2440 and identify three T6SS gene clusters (K1-, K2- and K3-T6SS). Besides, ten T6SS effector/immunity pairs were found, including putative nucleases and pore-forming colicins. We show that the K1-T6SS is a potent antibacterial device which secretes a toxic Rhs-type effector Tke2. Remarkably, P. putida eradicates a broad range of bacteria in a K1-T6SS-dependent manner, including resilient phytopathogens which demonstrates that the T6SS is instrumental to empower P. putida to fight against competitors. Furthermore, we observed a drastically reduced necrosis on the leaves of Nicotiana benthamiana during co-infection with P. putida and Xanthomonas campestris. Such protection is dependent on the activity of the P. putida T6SS. Many routes have been explored to develop biocontrol agents capable of manipulating the microbial composition of the rhizosphere and phyllosphere. Here we unveil a novel mechanism for plant biocontrol which needs to be considered for the selection of plant wardens whose mission is to prevent phytopathogen infections.
-
Journal articleJohnson R, Byrne A, Berger CN, et al., 2016,
The type III secretion system effector SptP of Salmonella enterica serovar Typhi
, Journal of Bacteriology, Vol: 199, ISSN: 1098-5530Strains of the various Salmonella enterica serovars cause gastroenteritis or typhoid fever in humans, with virulence depending on the action of two type III secretion systems (Salmonella pathogenicity island 1 [SPI-1] and SPI-2). SptP is a Salmonella SPI-1 effector, involved in mediating recovery of the host cytoskeleton postinfection. SptP requires a chaperone, SicP, for stability and secretion. SptP has 94% identity between S. enterica serovar Typhimurium and S Typhi; direct comparison of the protein sequences revealed that S Typhi SptP has numerous amino acid changes within its chaperone-binding domain. Subsequent comparison of ΔsptP S Typhi and S. Typhimurium strains demonstrated that, unlike SptP in S. Typhimurium, SptP in S Typhi was not involved in invasion or cytoskeletal recovery postinfection. Investigation of whether the observed amino acid changes within SptP of S Typhi affected its function revealed that S Typhi SptP was unable to complement S. Typhimurium ΔsptP due to an absence of secretion. We further demonstrated that while S. Typhimurium SptP is stable intracellularly within S Typhi, S Typhi SptP is unstable, although stability could be recovered following replacement of the chaperone-binding domain with that of S. Typhimurium. Direct assessment of the strength of the interaction between SptP and SicP of both serovars via bacterial two-hybrid analysis demonstrated that S Typhi SptP has a significantly weaker interaction with SicP than the equivalent proteins in S. Typhimurium. Taken together, our results suggest that changes within the chaperone-binding domain of SptP in S Typhi hinder binding to its chaperone, resulting in instability, preventing translocation, and therefore restricting the intracellular activity of this effector. IMPORTANCE: Studies investigating Salmonella pathogenesis typically rely on Salmonella Typhimurium, even though Salmonella Typhi causes the more severe disease in humans. As such, an understanding of S. Typhi
-
Journal articleValentini M, Laventie BJ, Moscoso JA, et al., 2016,
Correction: The Diguanylate Cyclase HsbD Intersects with the HptB Regulatory Cascade to Control Pseudomonas aeruginosa Biofilm and Motility.
, PLOS Genetics, Vol: 12, ISSN: 1553-7390 -
Journal articleValentini M, Laventie BJ, Moscoso J, et al., 2016,
The Diguanylate Cyclase HsbD Intersects with the HptB Regulatory Cascade to Control Pseudomonas aeruginosa Biofilm and Motility.
, PLOS Genetics, Vol: 12, ISSN: 1553-7390The molecular basis of second messenger signaling relies on an array of proteins that synthesize, degrade or bind the molecule to produce coherent functional outputs. Cyclic di-GMP (c-di-GMP) has emerged as a eubacterial nucleotide second messenger regulating a plethora of key behaviors, like the transition from planktonic cells to biofilm communities. The striking multiplicity of c-di-GMP control modules and regulated cellular functions raised the question of signaling specificity. Are c-di-GMP signaling routes exclusively dependent on a central hub or can they be locally administrated? In this study, we show an example of how c-di-GMP signaling gains output specificity in Pseudomonas aeruginosa. We observed the occurrence in P. aeruginosa of a c-di-GMP synthase gene, hsbD, in the proximity of the hptB and flagellar genes cluster. We show that the HptB pathway controls biofilm formation and motility by involving both HsbD and the anti-anti-sigma factor HsbA. The rewiring of c-di-GMP signaling into the HptB cascade relies on the original interaction between HsbD and HsbA and on the control of HsbD dynamic localization at the cell poles.
-
Journal articleAle A, Crepin VF, Collins, et al., 2016,
Model of host-pathogen Interaction dynamics links In vivo optical imaging and immune responses
, Infection and Immunity, Vol: 85, ISSN: 1098-5522Tracking disease progression in vivo is essential for the development of treatments against bacterial infection. Optical imaging has become a central tool for in vivo tracking of bacterial population development and therapeutic response. For a precise understanding of in vivo imaging results in terms of disease mechanisms derived from detailed postmortem observations, however, a link between the two is needed. Here, we develop a model that provides that link for the investigation of Citrobacter rodentium infection, a mouse model for enteropathogenic Escherichia coli (EPEC). We connect in vivo disease progression of C57BL/6 mice infected with bioluminescent bacteria, imaged using optical tomography and X-ray computed tomography, to postmortem measurements of colonic immune cell infiltration. We use the model to explore changes to both the host immune response and the bacteria and to evaluate the response to antibiotic treatment. The developed model serves as a novel tool for the identification and development of new therapeutic interventions.
-
Journal articleMacDonald J, Freemont PS, 2016,
Computational protein design with backbone plasticity
, Biochemical Society Transactions, Vol: 44, Pages: 1523-1529, ISSN: 1470-8752The computational algorithms used in the design of artificial proteins have become increasingly sophisticated in recent years, producing a series of remarkable successes. The most dramatic of these is the de novo design of artificial enzymes. The majority of these designs have reused naturally occurring protein structures as ‘scaffolds’ onto which novel functionality can be grafted without having to redesign the backbone structure. The incorporation of backbone flexibility into protein design is a much more computationally challenging problem due to the greatly increased search space, but promises to remove the limitations of reusing natural protein scaffolds. In this review, we outline the principles of computational protein design methods and discuss recent efforts to consider backbone plasticity in the design process.
-
Journal articlePollard DJ, Young JC, Covarelli V, et al., 2016,
The type III secretion system effector SeoC of Salmonella enterica subspecies salamae and arizonae ADP-ribosylates Src and inhibits opsono-phagocytosis
, Infection and Immunity, Vol: 84, Pages: 3618-3628, ISSN: 1098-5522Salmonella spp. utilize type III secretion systems (T3SS) to translocate effectors into the cytosol of mammalian host cells, subverting cell signaling and facilitating the onset of gastroenteritis. In this study we compared a draft genome assembly of S. enterica subsp. salamae strain 3588/07 (S. salamae) against the genomes of S. enterica subsp. enterica serovar Typhimurium strain LT2 and S. bongori strain 12419. S. salamae encode the Salmonella pathogenicity island (SPI)-1; SPI-2 and the locus of enterocyte effacement (LEE) T3SSs. Though several key S. Typhimurium effector genes are missing (e.g. avrA, sopB and sseL), S. salamae invades HeLa cells and contain homologues of S. bongori sboK and sboC, which we named seoC. SboC and SeoC are homologues of EspJ from enteropathogenic and enterohaemorrhagic E. coli (EPEC and EHEC), which inhibits Src kinase-dependent phagocytosis by ADP-ribosylation. By screening 73 clinical and environmental Salmonella isolates we identified EspJ homologues in S. bongori, S. salamae and S. enterica subsp. arizonae (S. arizonae). The β-lactamase TEM-1 reporter system showed that SeoC is translocated by the SPI-1 T3SS. All the Salmonella SeoC/SboC homologues ADP-ribosylate Src E310 in vitro. Ectopic expression of SeoC/SboC inhibited phagocytosis of IgG-opsonized bead into Cos-7 cells stably expressing GFP-FcγRIIa. Concurrently, S. salamae infection of J774.A1 macrophages inhibited phagocytosis of beads, in a seoC dependent manner. These results show that S. bongori, S. salamae and S. arizonae share features of the infection strategy of extracellular pathogens EPEC and EHEC and sheds light on the complexities of the T3SS effector repertoires of Enterobacteriaceae.
-
Journal articleMacDonald JT, Kabasakal BV, Godding D, et al., 2016,
Synthetic beta-solenoid proteins with the fragment-free computational design of a beta-hairpin extension
, Proceedings of the National Academy of Sciences of the United States of America, Vol: 113, Pages: 10346-10351, ISSN: 1091-6490The ability to design and construct structures with atomic level precisionis one of the key goals of nanotechnology. Proteins offer anattractive target for atomic design, as they can be synthesized chemicallyor biologically, and can self-assemble. However the generalizedprotein folding and design problem is unsolved. One approach tosimplifying the problem is to use a repetitive protein as a scaffold.Repeat proteins are intrinsically modular, and their folding and structuresare better understood than large globular domains. Here, wehave developed a new class of synthetic repeat protein, based onthe pentapeptide repeat family of beta-solenoid proteins. We haveconstructed length variants of the basic scaffold, and computationallydesigned de novo loops projecting from the scaffold core. Theexperimentally solved 3.56 ˚A resolution crystal structure of one designedloop matches closely the designed hairpin structure, showingthe computational design of a backbone extension onto a syntheticprotein core without the use of backbone fragments from knownstructures. Two other loop designs were not clearly resolved in thecrystal structures and one loop appeared to be in an incorrect conformation.We have also shown that the repeat unit can accommodatewhole domain insertions by inserting a domain into one of the designedloops.
-
Journal articleCrepin VF, Collins JW, Habibzay M, et al., 2016,
Citrobacter rodentium mouse model of bacterial infection.
, Nature Protocols, Vol: 11, Pages: 1851-1876, ISSN: 1754-2189Infection of mice with Citrobacter rodentium is a robust model to study bacterial pathogenesis, mucosal immunology, the health benefits of probiotics and the role of the microbiota during infection. C. rodentium was first isolated by Barthold from an outbreak of mouse diarrhea in Yale University in 1972 and was 'rediscovered' by Falkow and Schauer in 1993. Since then the use of the model has proliferated, and it is now the gold standard for studying virulence of the closely related human pathogens enteropathogenic and enterohemorrhagic Escherichia coli (EPEC and EHEC, respectively). Here we provide a detailed protocol for various applications of the model, including bacterial growth, site-directed mutagenesis, mouse inoculation (from cultured cells and after cohabitation), monitoring of bacterial colonization, tissue extraction and analysis, immune responses, probiotic treatment and microbiota analysis. The main protocol, from mouse infection to clearance and analysis of tissues and host responses, takes ∼5 weeks to complete.
-
Journal articleFurniss RCD, Slater S, Frankel G, et al., 2016,
Enterohaemorrhagic E. coli modulates an ARF6:Rab35 signalling axis to prevent recycling endosome maturation during infection
, Journal of Molecular Biology, Vol: 428, Pages: 3399-3407, ISSN: 1089-8638Enteropathogenic and enterohaemorrhagic Escherichia coli (EPEC/EHEC) manipulate a plethora of host cell processes to establish infection of the gut mucosa. This manipulation is achieved via the injection of bacterial effector proteins into host cells using a Type III secretion system. We have previously reported that the conserved EHEC and EPEC effector EspG disrupts recycling endosome function, reducing cell surface levels of host receptors through accumulation of recycling cargo within the host cell. Here we report that EspG interacts specifically with the small GTPases ARF6 and Rab35 during infection. These interactions target EspG to endosomes and prevent Rab35-mediated recycling of cargo to the host cell surface. Furthermore, we show that EspG has no effect on Rab35-mediated uncoating of newly formed endosomes, and instead leads to the formation of enlarged EspG/TfR/Rab11 positive, EEA1/Clathrin negative stalled recycling structures. Thus, this paper provides a molecular framework to explain how EspG disrupts recycling whilst also reporting the first known simultaneous targeting of ARF6 and Rab35 by a bacterial pathogen.
-
Journal articleSchuster C, Bellows L, Tosi T, et al., 2016,
The second messenger c-di-AMP inhibits the osmolyte uptake system OpuC in Staphylococcus aureus
, Science Signaling, Vol: 9, Pages: ra81-ra81, ISSN: 1945-0877Staphylococcus aureus is an important opportunistic human pathogen that is highly resistant to osmotic stresses. To survive an increase in osmolarity, bacteria immediately take up potassium ions and small organic compounds known as compatible solutes. The second messenger cyclic diadenosine monophosphate (c-di-AMP) reduces the ability of bacteria to withstand osmotic stress by binding to and inhibiting several proteins that promote potassium uptake. We identified OpuCA, the adenosine triphosphatase (ATPase) component of an uptake system for the compatible solute carnitine, as a c-di-AMP target protein in S. aureus and found that the LAC*ΔgdpP strain of S. aureus, which overproduces c-di-AMP, showed reduced carnitine uptake. The paired cystathionine-β-synthase (CBS) domains of OpuCA bound to c-di-AMP, and a crystal structure revealed a putative binding pocket for c-di-AMP in the cleft between the two CBS domains. Thus, c-di-AMP inhibits osmoprotection through multiple mechanisms.
-
Journal articleRasheed M, Garnett J, Perez-Dorado I, et al., 2016,
Crystal structure of the CupB6 adhesive tip from the chaperone-usher family of pili from Pseudomonas aeruginosa
, Biochimica et Biophysica Acta - Protein Structure, Vol: 1864, Pages: 1500-1505, ISSN: 0005-2795Pseudomonas aeruginosa is a Gram-negative opportunistic bacterial pathogen that can cause chronicinfection of the lungs of cystic fibrosis patients. Chaperone-usher systems in P. aeruginosa are knownto translocate and assemble adhesive pili on the bacterial surface and contribute to biofilm formationwithin the host. Here, we report the crystal structure of the tip adhesion subunit CupB6 from thecupB1-6 gene cluster. The tip domain is connected to the pilus via the N-terminal donor strand fromthe main pilus subunit CupB1. Although the CupB6 adhesion domain bears structural features similarto other CU adhesins it displays an unusual polyproline helix adjacent to a prominent surface pocket,which are likely the site for receptor recognition.
-
Journal articleFilloux A, Freemont P, 2016,
Structural biology: baseplates in contractile machines
, Nature Microbiology, Vol: 1, ISSN: 2058-5276 -
Journal articlePlanamente S, Salih O, Manoli E, et al., 2016,
TssA forms a gp6-like ring attached to the type VI secretion sheath
, EMBO Journal, Vol: 35, Pages: 1613-1627, ISSN: 0261-4189The type VI secretion system (T6SS) is a supra-molecular bacterial complex that resembles phage tails. It is a killing machine which fires toxins into target cells upon contraction of its TssBC sheath. Here, we show that TssA1 is a T6SS component forming dodecameric ring structures whose dimensions match those of the TssBC sheath and which can accommodate the inner Hcp tube. The TssA1 ring complex binds the T6SS sheath and impacts its behaviour in vivo. In the phage, the first disc of the gp18 sheath sits on a baseplate wherein gp6 is a dodecameric ring. We found remarkable sequence and structural similarities between TssA1 and gp6 C-termini, and propose that TssA1 could be a baseplate component of the T6SS. Furthermore, we identified similarities between TssK1 and gp8, the former interacting with TssA1 while the latter is found in the outer radius of the gp6 ring. These observations, combined with similarities between TssF and gp6N-terminus or TssG and gp53, lead us to propose a comparative model between the phage baseplate and the T6SS.
-
Journal articleHolmes AH, Gill SK, Hui K, et al., 2016,
Increased airway glucose increases airway bacterial load in hyperglycaemia
, Scientific Reports, Vol: 6, ISSN: 2045-2322Diabetes is associated with increased frequency of hospitalization due to bacterial lung infection.We hypothesize that increased airway glucose caused by hyperglycaemia leads to increasedbacterial loads. In critical care patients, we observed that respiratory tract bacterial colonisationis significantly more likely when blood glucose is high. We engineered mutants in genesaffecting glucose uptake and metabolism (oprB, gltK, gtrS and glk) in Pseudomonas aeruginosa,strain PAO1. These mutants displayed attenuated growth in minimal medium supplemented withglucose as the sole carbon source. The effect of glucose on growth in vivo was tested usingstreptozocin-induced, hyperglycaemic mice, which have significantly greater airway glucose.Bacterial burden in hyperglycaemic animals was greater than control animals when infected withwild type but not mutant PAO1. Metformin pre-treatment of hyperglycaemic animals reducedboth airway glucose and bacterial load. These data support airway glucose as a criticaldeterminant of increased bacterial load during diabetes.
-
Journal articleLund-Palau H, Turnbull AR, Bush A, et al., 2016,
Pseudomonas aeruginosa infection in cystic fibrosis: pathophysiological mechanisms and therapeutic approaches
, Expert Review of Respiratory Medicine, Vol: 10, Pages: 685-697, ISSN: 1747-6348Pseudomonas aeruginosa is a remarkably versatile environmental bacterium with an extraordinary capacity to infect the cystic fibrosis (CF) lung. Infection with P. aeruginosa occurs early, and although eradication can be achieved following early detection, chronic infection occurs in over 60% of adults with CF. Chronic infection is associated with accelerated disease progression and increased mortality. Extensive research has revealed complex mechanisms by which P. aeruginosa adapts to and persists within the CF airway. Yet knowledge gaps remain, and prevention and treatment strategies are limited by the lack of sensitive detection methods and by a narrow armoury of antibiotics. Further developments in this field are urgently needed in order to improve morbidity and mortality in people with CF. Here, we summarize current knowledge of pathophysiological mechanisms underlying P. aeruginosa infection in CF. Established treatments are discussed, and an overview is offered of novel detection methods and therapeutic strategies in development.
-
Journal articleChambonnier G, Roux L, Redelberger D, et al., 2016,
The Hybrid Histidine Kinase LadS Forms a Multicomponent Signal Transduction System with the GacS/GacA Two-Component System in Pseudomonas aeruginosa.
, PLOS Genetics, Vol: 12, ISSN: 1553-7390In response to environmental changes, Pseudomonas aeruginosa is able to switch from a planktonic (free swimming) to a sessile (biofilm) lifestyle. The two-component system (TCS) GacS/GacA activates the production of two small non-coding RNAs, RsmY and RsmZ, but four histidine kinases (HKs), RetS, GacS, LadS and PA1611, are instrumental in this process. RetS hybrid HK blocks GacS unorthodox HK autophosphorylation through the formation of a heterodimer. PA1611 hybrid HK, which is structurally related to GacS, interacts with RetS in P. aeruginosa in a very similar manner to GacS. LadS hybrid HK phenotypically antagonizes the function of RetS by a mechanism that has never been investigated. The four sensors are found in most Pseudomonas species but their characteristics and mode of signaling may differ from one species to another. Here, we demonstrated in P. aeruginosa that LadS controls both rsmY and rsmZ gene expression and that this regulation occurs through the GacS/GacA TCS. We additionally evidenced that in contrast to RetS, LadS signals through GacS/GacA without forming heterodimers, either with GacS or with RetS. Instead, we demonstrated that LadS is involved in a genuine phosphorelay, which requires both transmitter and receiver LadS domains. LadS signaling ultimately requires the alternative histidine-phosphotransfer domain of GacS, which is here used as an Hpt relay by the hybrid kinase. LadS HK thus forms, with the GacS/GacA TCS, a multicomponent signal transduction system with an original phosphorelay cascade, i.e. H1LadS→D1LadS→H2GacS→D2GacA. This highlights an original strategy in which a unique output, i.e. the modulation of sRNA levels, is controlled by a complex multi-sensing network to fine-tune an adapted biofilm and virulence response.
-
Journal articleSawicka M, Wanrooij PH, Darbari VC, et al., 2016,
The dimeric architecture of checkpoint kinases Mec1ATR and Tel1ATM reveal a common structural organisation.
, Journal of Biological Chemistry, Vol: 291, Pages: 13436-13447, ISSN: 1083-351XThe phosphatidylinositol 3-kinase-related protein kinases (PIKKs) are key regulators controlling a wide range of cellular events. The yeast Tel1 and Mec1-Ddc2 complex (ATM and ATR-ATRIP in humans) play pivotal roles in DNA replication, DNA damage signalling and repair. Here, we present the first structural insight for dimers of Mec1-Ddc2 and Tel1 using single particle electron microscopy. Both kinases reveal a head-to-head dimer with one major dimeric interface through their N-terminal HEAT repeats. Their dimeric interface is significantly distinct from the interface of mTOR Complex 1 dimer, which oligomerises through two spatially separate interfaces. We also observe different structural organisation of kinase domains of Mec1 and Tel1. The kinase domains in the Mec1-Ddc2 dimer are located in close proximity to each other. However, in the Tel1 dimer they are fully separated providing potential access of substrates to this kinase, even in its dimeric form.
-
Journal articleTaylor JD, Hawthorne WJ, Lo J, et al., 2016,
Electrostatically-guided inhibition of Curli amyloid nucleation by the CsgC-like family of chaperones
, Scientific Reports, Vol: 6, ISSN: 2045-2322Polypeptide aggregation into amyloid is linked with several debilitating human diseases.Despite the inherent risk of aggregation-induced cytotoxicity, bacteria control the export ofamyloid-prone subunits and assemble adhesive amyloid fibres during biofilm formation. AnEscherichia protein, CsgC potently inhibits amyloid formation of curli amyloid proteins.Here we unlock its mechanism of action, and show that CsgC strongly inhibits primarynucleation via electrostatically-guided molecular encounters, which expands theconformational distribution of disordered curli subunits. This delays the formation of higherorder intermediates and maintains amyloidogenic subunits in a secretion-competent form.New structural insight also reveal that CsgC is part of diverse family of bacterial amyloidinhibitors. Curli assembly is therefore not only arrested in the periplasm, but the preservationof conformational flexibility also enables efficient secretion to the cellsurface. Understanding how bacteria safely handle amyloidogenic polypeptides contributetowards efforts to control aggregation in disease-causing amyloids and amyloid-based biotechnological applications.
-
Journal articleValentini M, Filloux A, 2016,
Biofilms and cyclic di-GMP (c-di-GMP) signaling: lessons from Pseudomonas aeruginosa and other bacteria
, Journal of Biological Chemistry, Vol: 291, Pages: 12547-12555, ISSN: 1083-351XThe cyclic-di-GMP (c-di-GMP) second messenger represents a signaling system that regulates many bacterial behaviors and is of key importance for driving the lifestyle switch between motile loner cells and biofilm formers. This review provides an up-to-date compendium of c-di-GMP pathways connected to biofilm formation, biofilm-associated motilities and other functionalities in the ubiquitous and opportunistic human pathogen Pseudomonas aeruginosa. This bacterium is frequently adopted as model organism to study bacterial biofilm formation. Importantly, its versatility and adaptation capabilities are linked with a broad range of complex regulatory networks, including a large set of genes involved in c-di-GMP biosynthesis, degradation and transmission.
-
Journal articleMiliara X, Matthews S, 2016,
Structural comparison of yeast and human intra-mitochondrial lipid transport systems
, Biochemical Society Transactions, Vol: 44, Pages: 479-485, ISSN: 1470-8752Mitochondria depend on a tightly regulated supply of phospholipids. The protein of relevant evolutionary and lymphoid interest (PRELI)/Ups1 family together with its mitochondrial chaperones [TP53-regulated inhibitor of apoptosis 1 (TRIAP1)/Mdm35] represents a unique heterodimeric lipid-transfer system that is evolutionary conserved from yeast to man. Recent X-ray crystal structures of the human and yeast systems are compared and discuss here and shed new insight into the mechanism of the PRELI/Ups1 system.
-
Journal articleSo EC, Schroeder GN, Carson D, et al., 2016,
The Rab-binding profiles of bacterial virulence factors during infection
, Journal of Biological Chemistry, Vol: 291, Pages: 5832-5843, ISSN: 1083-351XLegionella pneumophila, the causativeagent of Legionnaire’s disease, uses its typeIV secretion system to translocate over 300effector proteins into host cells. Theseeffectors subvert host cell signalingpathways to ensure bacterial proliferation.Despite their importance for pathogenesis,the roles of most of the effectors are yet tobe characterized. Key to understanding thefunction of effectors is the identification ofhost proteins they bind during infection. Wepreviously developed a novel tandemaffinitypurification (TAP) approach usinghexahistidine and BirA-specificbiotinylation tags for isolating translocatedeffector complexes from infected cellswhose composition were subsequentlydeciphered by mass spectrometry. Here wefurther advanced the workflow for the TAPapproach and determined the infectiondependentinteractomes of the effectorsSidM and LidA, which were previouslyreported to promiscuously bind multiple RabGTPases in vitro. In this study we defined astringent subset of Rab GTPases targeted bySidM and LidA during infection, comprisingof Rab1A, 1B, 6 and 10; in addition, LidAtargets Rab14 and 18. Taken together, thisstudy illustrates the power of this approachto profile the intracellular interactomes ofbacterial effectors during infection
-
Journal articleDomingues L, Ismail A, Charro N, et al., 2016,
The Salmonella effector SteA binds phosphatidylinositol 4-phosphate for subcellular targeting within host cells
, Cellular Microbiology, Vol: 18, Pages: 949-969, ISSN: 1462-5822Many bacterial pathogens use specialized secretion systems to deliver virulence effector proteins into eukaryotic host cells. The function of these effectors depends on their localization within infected cells, but the mechanisms determining subcellular targeting of each effector are mostly elusive. Here, we show that the Salmonella type III secretion effector SteA binds specifically to phosphatidylinositol 4-phosphate [PI(4)P]. Ectopically expressed SteA localized at the plasma membrane (PM) of eukaryotic cells. However, SteA was displaced from the PM of Saccharomyces cerevisiae in mutants unable to synthesize the local pool of PI(4)P and from the PM of HeLa cells after localized depletion of PI(4)P. Moreover, in infected cells, bacterially translocated or ectopically expressed SteA localized at the membrane of the Salmonella-containing vacuole (SCV) and to Salmonella-induced tubules; using the PI(4)P-binding domain of the Legionella type IV secretion effector SidC as probe, we found PI(4)P at the SCV membrane and associated tubules throughout Salmonella infection of HeLa cells. Both binding of SteA to PI(4)P and the subcellular localization of ectopically expressed or bacterially translocated SteA were dependent on a lysine residue near the N-terminus of the protein. Overall, this indicates that binding of SteA to PI(4)P is necessary for its localization within host cells.
-
Journal articleFilloux A, Whitfield C, 2016,
Editorial: The many wonders of the bacterial cell surface
, FEMS MICROBIOLOGY REVIEWS, Vol: 40, Pages: 161-163, ISSN: 0168-6445- Author Web Link
- Cite
- Citations: 9
-
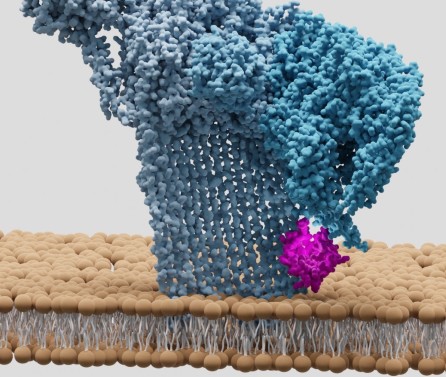
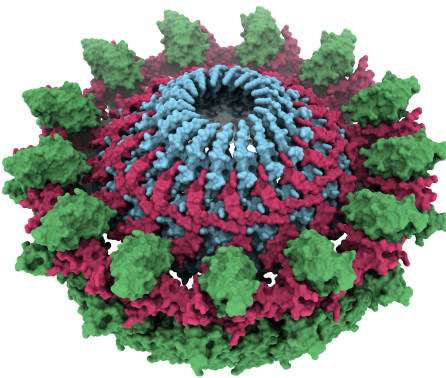
Journal articleSerna Gil M, Bubeck D, Giles JL, et al., 2016,
Structural basis of complement membrane attack complex formation
, Nature Communications, Vol: 7, Pages: 1-7, ISSN: 2041-1723In response to complement activation, the membrane attack complex (MAC) assembles from fluid-phase proteins to form pores in lipid bilayers. MAC directly lyses pathogens by a ‘multi-hit’ mechanism; however, sublytic MAC pores on host cells activate signalling pathways. Previous studies have described the structures of individual MAC components and subcomplexes; however, the molecular details of its assembly and mechanism of action remain unresolved. Here we report the electron cryo-microscopy structure of human MAC at subnanometre resolution. Structural analyses define the stoichiometry of the complete pore and identify a network of interaction interfaces that determine its assembly mechanism. MAC adopts a ‘split-washer’ configuration, in contrast to the predicted closed ring observed for perforin and cholesterol-dependent cytolysins. Assembly precursors partially penetrate the lipid bilayer, resulting in an irregular β-barrel pore. Our results demonstrate how differences in symmetric and asymmetric components of the MAC underpin a molecular basis for pore formation and suggest a mechanism of action that extends beyond membrane penetration.
This data is extracted from the Web of Science and reproduced under a licence from Thomson Reuters. You may not copy or re-distribute this data in whole or in part without the written consent of the Science business of Thomson Reuters.
Centre for Structural Biology Open Day
Join us for our Open Day on 16 May 2024 - find out more!