A simple, general and computationally efficient method for computing optical spectra
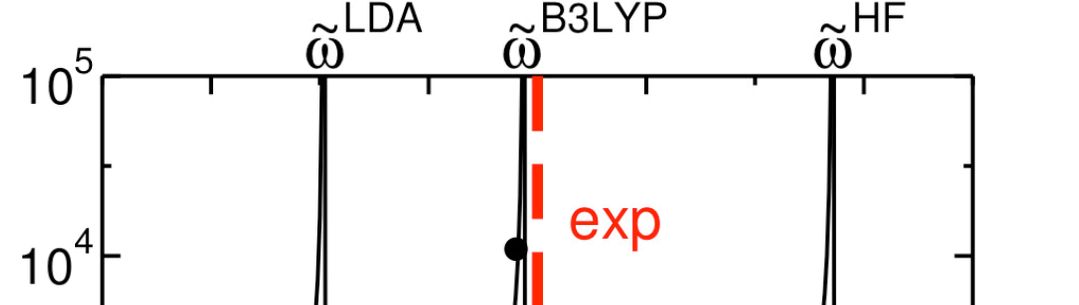
Time dependent density functional theory provides a rigorous basis for the prediction of optical gaps. The accuracy of the calculated optical spectrum is however limited by our ability to describe the spatially and temporally delocalised many body response of the electrons to an applied field, that is, to find a suitable approximation to the TDDFT response kernel. We have recently implemented a simple, general and computationally convenient approach to TD-DFT for periodic and finite systems in the all electron CRYSTAL code. Approximating the response kernel with non-local hybrid exchange functionals is found to be essential for reproducing the optical properties of a wide variety of direct and indirect gap systems to within experimental accuracy.
Computational Materials Science
